Speakers - Symposium 2022
Table of contents
Prof. Sven Rau

CataLight / University of Ulm, DE
Sven Rau studied Chemistry at Friedrich-Schiller-University Jena and at Dublin City University in Ireland. He received his PhD in 2000 and later, in 2007, his habilitation from FSU Jena. He became a professor at the Friedrich-Alexander-University Erlangen-Nuremberg in 2008. Since 2011 he is a full professor at Ulm University and is spokesperson of the collaborative research centre 234 – CataLight since 2018.
Sven's research interests include artificial photosynthesis and photodynamic therapy. At the centre of this are photoactive assemblies based on transition metal complexes. The Rau group has a strong expertise in designing and synthesising photoactive assemblies and investigates their performance in light-driven catalysis or their photochemical and photophysical behaviour in different environments.
Repairing efficient light driven catalysts for hydrogen generation
Photocatalytic water splitting can be utilised with molecular photocatalysts. Ruthenium complexes with tpphz and related type bridging ligands play an important role within this field. [1,2] Through a series of model catalysts we now can differentiate between several important structural aspects influencing the photocatalytic activity.
One very limiting factor for applying molecular catalysts is their propensity to loss of activity. Nature has devised elaborate repair systems restoring catalytic activity of biological molecular defined catalysts. [3] We present here a new concept on repairing the below described molecular photocatalyst and thereby boosting catalytic performance.
-
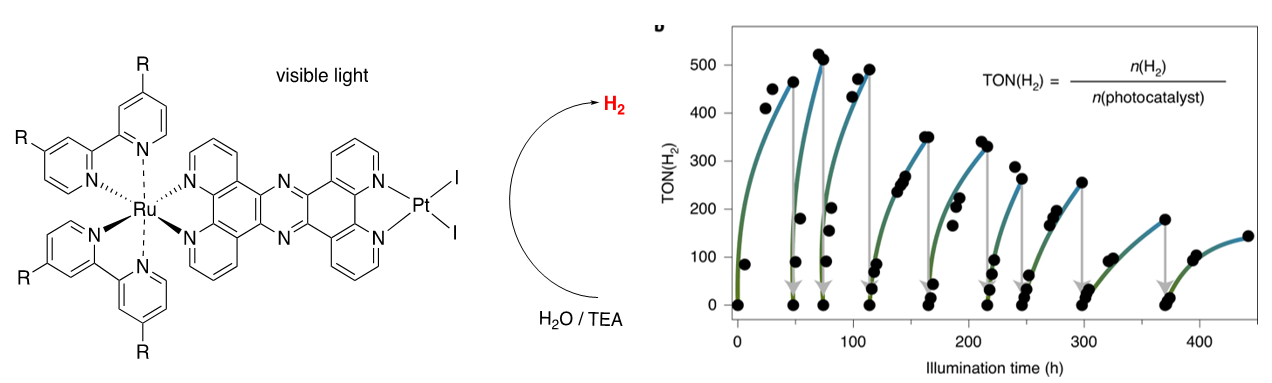
- Photocatalyst for hydrogen production which can be repaired.
Dr. Gabriele Colombo

Casale SA, CH
Gabriele Colombo studied chemical engineering at ETH Zürich. He then completed his PhD in Materials Science at ETH in 2019, in the group of Soft Materials of Jan Vermant. The PhD focused on the characterization of colloidal dispersions and colloidal gels by a combination of confocal microscopy and rheometry.
He joined Casale SA as R&D Technology engineer in 2019, where he mainly works on the development of innovative production processes for ammonia, methanol and nitric acid, targeting improved energy efficiency and sustainable production.
Decarbonization of Hydrogen and Ammonia Production Processes in Industry
Hydrogen is a key feedstock for the chemical industry, where it is used in large amounts in petrochemical refining processes and in the production of methanol and ammonia. Ammonia represents the backbone of the fertilizers product tree, where ammonium nitrate and urea are the most traded derivatives.
The current production of hydrogen is located at the points of use and relies almost entirely on fossil feedstock, for the large majority natural gas. The state-of-the-art process utilizes the strongly endothermic methane steam reforming reaction, yielding a mixture of mostly hydrogen and carbon monoxide called synthesis gas. The carbon monoxide is further converted to hydrogen by water gas shift reactions and the resulting CO2 is removed from the product gas and vented to the atmosphere. As a result, hydrogen production is an energy intensive process that accounts for 2% of the total global primary energy demand, generating correspondingly significant CO2 emissions (10 kgCO2/kgH2).
In addition to current uses, that will remain necessary in the chemical industry, hydrogen is expected to be needed for the energy system of the future as a carbon-free energy carrier. The pre-requisite for such use is of course a production route with no CO2 emissions, or at least strongly decreased emissions with respect to steam reforming.
Mature technologies are already available to produce low-carbon hydrogen through either water electrolysis (green hydrogen) or steam reforming followed by CO2 capture and permanent underground sequestration (blue hydrogen/CCS). However, green hydrogen is currently produced at a cost about 3–8 times larger than with methane reforming, and scale-up of water electrolyzers is still under way. Blue hydrogen is already costcompetitive but constrained by the availability of CO2 sequestration capacity and CO2 transport infrastructure. New, more cost-efficient routes of low-carbon hydrogen production are therefore still desirable as successors of water electrolysis and CCS, to bring forward the development of the hydrogen
infrastructure.
Prof. Jenny Y. Yang

University of California, Irvine, USA
Jenny Y. Yang received her B.S. at UC Berkeley (research with Professor Jeffrey R. Long) and completed her doctoral studies at MIT under the guidance of Professor Daniel G. Nocera. After her postdoctoral position with Dr. Daniel L. Dubois at the Pacific Northwest National Laboratory, she was hired as a research scientist in the Center for Molecular Electrocatalysis. After a subsequent position as a scientist at the Joint Center for Artificial Photosynthesis, she started her current position as a faculty member at the University of California, Irvine in 2013. She was promoted to Associate Professor in 2019 and Professor in 2021.
She is currently an Associate Editor for the Journal of the American Chemical Society and Director of the Center for Closing the Carbon Cycle (4C), and Energy Frontier Research Center.
Electrochemical CO2 Capture, Concentration, and Conversion
Closing the anthropogenic carbon cycle requires efficient methods for capture and concentration from dilute streams. Current methods for CCC are energy intensive due to their reliance on thermal cycles, which are intrinsically Carnot limited in efficiency. In contrast, electrochemically driven CCC (eCCC) can operate at much higher theoretical efficiencies. However, most reported systems are sensitive to O2, precluding their practical use. Prior efforts to chemically modify redox carriers to operate at milder potentials resulted in a loss in CO2 binding. To overcome these limitations, we used common alcohols additives to anodically shift the reduction potential of a quinone redox carrier, 2,3,5,6-tetrachloro-p-benzoquinone (TCQ), by up to 350 mV, conferring O2 stability. We demonstrate a full cycle of eCCC in aerobic simulated flue gas using TCQ and ethanol, two commercially available compounds. Based on the system properties, an estimated minimum of 21 kJ/mol is required to concentrate CO2 from 10% to 100%, or twice as efficient as state-of-the-art thermal amine capture systems and other reported redox carrier-based systems. Furthermore, this approach of using hydrogen-bond donor additives is general and can be used to tailor the redox properties of other quinones/alcohol combinations for specific CO2 capture applications.
Prof. Theodor Agapie

California Institute of Technology, USA
Originally from Bucharest, Romania, Agapie received his B.S. degree from Massachusetts Institute of Technology in 2001 and his Ph.D. from California Institute of Technology in 2007. He was a Miller Postdoctoral Fellow at University of California, Berkeley. Agapie returned to Caltech in 2009 to start his independent career as Assistant Professor of Chemistry. He has been Professor of Chemistry since 2014.
His research focuses on synthetic inorganic and organometallic chemistry on topics related to energy conversion, catalysis, and upgrading readily available chemical precursors. He has ~120 scientific publications and has presented over 100 invited lectures at international and US universities, companies, and conferences. Selected awards include the ACS Award in Pure Chemistry (2013), Presidential Early Career Award for Scientists and Engineers (PECASE, 2014), and Humboldt Foundation’s Bessel Research Award (2022).
Electrocatalytic CO2 Reduction to Multicarbon Products with Hybrid Cu-Organic Electrodes
Toward utilizing CO2 as a carbon source for the generation of liquid fuels and other value-added chemicals using renewable energy, the development of chemical transformations for the preparation of multicarbon products is desirable. Electrochemical CO2 reduction (CO2R) on Cu electrodes generates a mixture of products, including CO, methane, ethylene, ethanol, and propanol, and it competes with hydrogen evolution. To improve the selectivity of CO2R, hybrid electrodes based on Cu coated with an organic layer were prepared targeting microenvironments that facilitate the formation of C2+ products. Addition of N-aryl pyridinium in the electrolyte results in almost complete suppression of methane formation and decrease in hydrogen generation in CO2R with Cu, for a combined selectivity for C2 and C3 products of ~80%. The formation of an organic film consisting of the product of reductive dimerization of N-aryl pyridinium is required for improved selectivity of CO2R to multicarbon products. A variety of organic precursors have been explored to optimize the organic film generation and its impact on CO2R performance with Cu and alloys. Mechanistic implications will be discussed.
Dr. Aaron Appel

Pacific Northwest National Laboratory,USA
Aaron Appel is a chemist and Team Leader for Molecular Catalysis in the Catalysis Science Group at PNNL. His research focuses on designing catalysts and electro-catalysts for interconversion of energy and fuels, as well as electrochemically switchable sorbents for CO2. His research has two aspects: thermodynamic studies of discreet bond forming and bond breaking events, and the design and testing of complexes and catalysts based on the resulting thermodynamic data. Aaron has applied this approach to the production and utilization of hydrogen, to the reduction of carbon dioxide to liquid fuels, and to transformations of additional small molecules for energy storage.
Aaron holds a PhD in Chemistry from the University of Colorado Boulder and a BS in Chemistry from Washington State University.
Design of Molecular Catalysts for Hydride Transfer Reactions and Electrocatalytic Oxidation of Alcohols
Thermodynamic parameters are invaluable for the design of molecular catalysts for energy storage reactions. They can provide a wealth of information, including the standard potentials for reactions, the energies for discrete bond forming and bond breaking events, and quantitative information about the impacts of solvents and reaction conditions, all of which can be used for the design of catalytic systems.
Alcohols are potential liquid solar fuels that enable large-scale energy storage, but their efficient use is critical to an effective energy storage cycle. Electrocatalytic oxidation of alcohols is an essential aspect of this fuel cycle, which would enable high efficiency retrieval of the stored energy. Relatively few molecular electrocatalysts have been reported for the oxidation of alcohols, and even fewer are based on non-precious metals. To compare the relative performances of reported electrocatalysts, the thermodynamic potentials under catalytic conditions have been estimated, and thereby overpotentials for reported catalysts have been determined. Amongst the reported electrocatalysts, (PtBu2NtBu2)Ni(MeCN)xn+ has been shown to be an effective catalyst for the oxidation of alcohols, with competitive rates and the lowest overpotential reported for a soluble molecular electrocatalyst for the oxidation of alcohols. Mechanistic studies of these catalysts have been recently reported, and now this system have been extended to include catalysis with monophosphines as additives, which greatly enhance catalytic rates and enable more challenging oxidations, including methanol.
(remote talk)
Dr. Yonggui Zhao

LightChEC / University of Zurich, CH
Yonggui Zhao received his BSc in electrical engineering from Northeastern University (China, 2014) and his MSc in solid-state electronics from Tianjin University (China, 2017). Subsequently, he worked as a junior researcher in experimental physics at the University of Amsterdam. He then joined the group of Prof. Greta R. Patzke at the University of Zurich as a PhD candidate in Dec. 2017 and finished his PhD thesis in May 2022.
Currently, he is a postdoctoral researcher with the URPP LightChEC at UZH. His main research interest is in situ monitoring and understanding of water splitting electrocatalysts under operational conditions.
Operando spectroscopic unveiling of the structural motif dynamics in NiFe-based coordination polymers for water oxidation
Water splitting into hydrogen and oxygen is one of the most promising strategies for renewable energy conversion. [1] However, the demanding four-electron transfer reaction, namely water oxidation reaction (OER), renders efficient production of hydrogen challenging. During the past decades, great progress has been made in the development of cost-efficient OER catalysts. Among these catalysts, NiFe-based coordination polymers (CP) have attracted much interest due to their unique structural properties, namely tunable metal centers and high surface areas. [2,3]
Recently, our group reported on an ultra-thin NiFe-CPs for OER, followed by a room-temperature reduction strategy for the preparation of the low-crystalline reduced NiFe-CPs (R-NiFe-CPs). [3] Spectroscopic studies evidenced that the as-prepared R-NiFe-CPs contained rich structural vacancies, namely oxygen and nickel vacancies. Density functional theory (DFT) calculations revealed that the presence of structural deficiencies facilitated the adsorption/desorption of OER intermediates and therefore, the as-prepared R-NiFe-CPs showed an excellent OER activity. However, a key issue, as to how do the introduced structural vacancies dynamically alter the formation of active species and the O-O bond coupling pathway? remains to be resolved. With the help of operando X-ray absorption spectroscopy (XAS) monitoring, we found that the OER of R-NiFe-CPs was hindered by the construction of active NiIV-O-FeIV moieties. Engineering S heteroanion incorporation into R-NiFe-CPs (referred to as S-R-NiFe-CPs) not only promoted the formation of OER active S-NiIVO-FeIVO moieties but also lowered the energy barrier for O-O bond formation. Due to the optimized OER kinetics, the as-prepared S-R-NiFe-CPs delivered a high OER activity with a very low overpotential of 235 mV at 10 mA/cm2, outperforming most recently reported NiFe-based OER electrocatalysts.