Electronic Structure Calculations of Molecules at Interfaces
Lead by Prof. Jürg Hutter and Dr. Marcella Iannuzzi
Molecular systems anchored at metal and oxide surfaces have great potential for applications in dye-sensitized solar cells for electric power generation and in dye-sensitized photoelectrochemical cells for energy conversion and storage. Experiments on these highly complex systems greatly profit from support by atomistic simulations. The interplay of experiment and simulation leads to improved interpretation and new insight in a synergetic way.
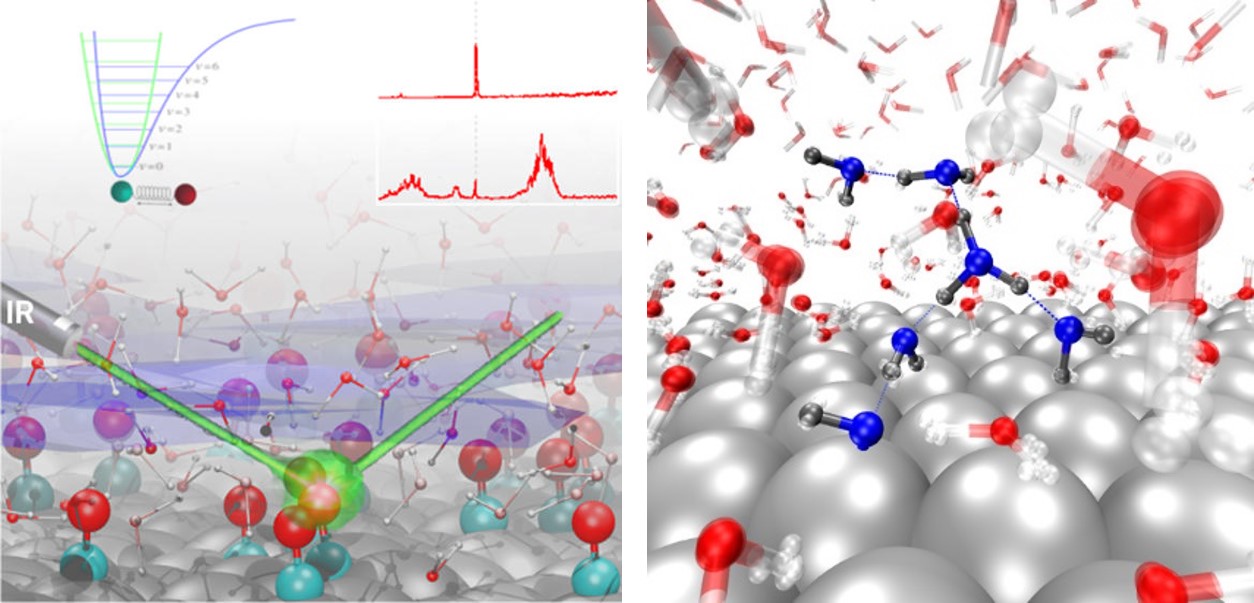
Realistic atomistic models of such complex systems need to describe the electronic structure of thousands of dynamic particles in an accurate manner. We are using density functional theory based methods as implemented in the CP2K software. CP2K allows ab initio molecular dynamics simulations of large systems using periodic boundary conditions and is making efficient use of modern supercomputers.
Our recent projects include:
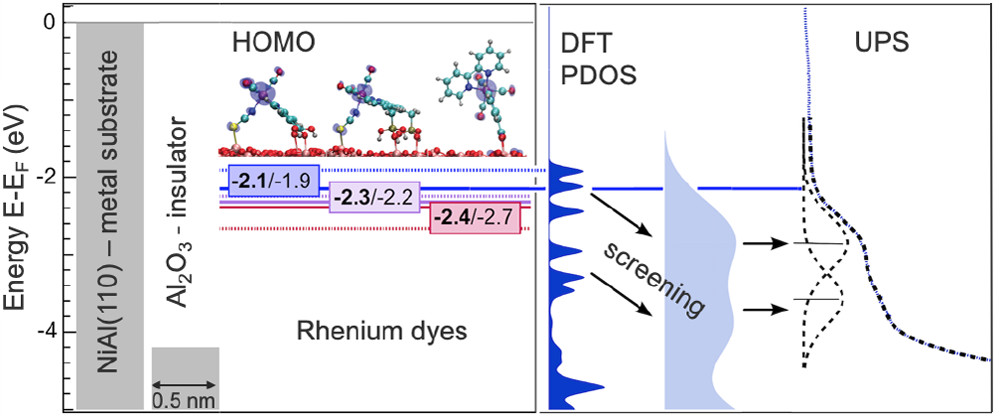
- Study of rhenium and ruthenium organometallic dyes anchored on alumina. DFT calculations for the Re dyes show that the most stable adsorption configurations exhibit bonding via the dedicated anchoring groups, with an additional sulfur-aluminum bond for the dyes containing a thiocyanate ligand. The alignment of the occupied molecular levels with respect to the alumina valence band maximum, follows the experimental trend. Dynamical charge screening is found to be important for the systems when comparing UPS and DFT results.
W.-D. Zabka et al., J. Phys. Chem. C 2019, 123, 22250 - Ionization of Water as an Effect of Quantum Delocalization at Aqueous Electrode Interfaces. The enhanced probability of water dissociation at the aqueous platinum electrode interfaces is predicted by path-integral ab initio molecular dynamics. We characterize the dissociation mechanism and the dynamics of the formed water ions within the contact layer at the metal/water interface. Other metallic surface and the effect of an applied bias are going to be considered next.
J.Lan et al., J. Phys. Chem. Lett. 2020, 11, 3724 - Stable and tunable phosphonic acid dipole layer for band edge engineering of photoelectrochemical Photoelectrochemical (PEC) cells are operated in direct contact with corrosive electrolytes and measures have to be taken to protect the otherwise unstable materials. We studied a general method to tune the band alignment between photoabsorbers and TiO2, which makes otherwise unfavorable material combinations more efficient. The study is then extended to other oxides and other types of protective layers.
Rene Wick-Joliat et al. Energy Environ. Sci., 2019, 12, 1901 - Comparison of Penta and Tetra-pyridyl Cobalt-based Catalysts for Water Reduction: H2 Production Cycle, Solvent Response and Reduction Free Energy. The dynamic properties of the systems, including solvent response and structural changes occurring in the catalysts, are investigated by running ab initio molecular dynamics for all intermediate steps of the H2 production cycle. The reduction free energy for electron injection, and the related reorganisation energy are also estimated applying the thermodynamic integration scheme within the generalised Marcus theory.
Y. Gurdal and M. Iannuzzi, ChemPhysChem 2020, 21, 1. R. Alberto et al., Chimia 73 (2019) 906 - Host-guest interactions on electrode surfaces for immobilization of molecular catalysts. Anchoring molecular catalysts on electrode surfaces combines the high selectivity and activity of molecular systems with the practicality of heterogeneous systems. We investigate the reversible binding of molecular electrocatalysts to electrode surfaces via host-guest complexation with surface-anchored cyclodextrins. The host-guest interaction is remarkably strong and allows the flow of electrons between the electrode and the guest catalyst.
L. Severy et al, https://doi.org/10.26434/chemrxiv.12115026.v1